


監(jiān)管機構(gòu)
美國食品藥品監(jiān)督管理局(FDA)負責(zé)醫(yī)療器械的注冊,確保其安全性、有效性和創(chuàng)新性。
主要法規(guī)
聯(lián)邦食品、藥品和化妝品法案(FD&C Act)
21 CFR(Title 21- Code of Federal Regulations Parts 1-58. 800-1299)
國際醫(yī)療器械監(jiān)管機構(gòu)論壇(IMDRF)指南
美國國家標準協(xié)會(ANSI)標準
風(fēng)險分類
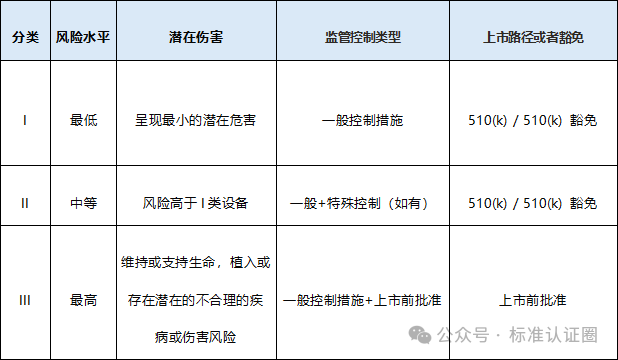
符合性評
-510(k):適用于大多數(shù)醫(yī)療器械的上市前通知。
-PMA(Premarket Approval):適用于高風(fēng)險醫(yī)療器械的上市前許可。
-De Novo 分類:適用于新類型的低到中等風(fēng)險醫(yī)療器械。
Premarket Notification 510(K)
510k 是在醫(yī)療器械產(chǎn)品進入美國市場之前提交給 FDA 的上市前技術(shù)文件,以證明該產(chǎn)品在預(yù)期用途、設(shè)計、材料、實施標準等方面與已經(jīng)合法銷售的同類產(chǎn)品具有相同的安全性和有效性。由于源自《聯(lián)邦食品、藥品和化妝品法案》的第 510 章的第 k 條款,因此被稱作 510k。
流程

提交510(k)申請的主要目的是證明該設(shè)備與已獲得FDA批準并在商業(yè)銷售的同品種設(shè)備 "實質(zhì)性等同"。FDA會對提交的申請進行審查,以確認這種等同性,并評估該設(shè)備是否按預(yù)期運行。
一旦FDA確認與原產(chǎn)品具有實質(zhì)等同性,他們就會簽發(fā)通過信(clearance letter),允許制造商在美國市場銷售該設(shè)備。值得注意的是,510(k)途徑下的許可并不意味著美國FDA的批準,而是表明該器械與現(xiàn)有市場器械的相似性。
FDA 510(K)產(chǎn)品注冊內(nèi)容
1) 申報資料,此部分應(yīng)包括申請人(或聯(lián)系人)和企業(yè)的基本信息、510(K)遞交的目的、申請上市器械的名稱型號和分類資料、產(chǎn)品代碼、進行實質(zhì)等效比較的產(chǎn)品(Predicate Device)名稱及其510(K)號碼;
2) 資料目錄,即510(K)文件中所含全部資料的清單(包括附件);
3) 真實性保證聲明,對此聲明,F(xiàn)DA有一個標準的樣本;
4) 產(chǎn)品名稱,即產(chǎn)品通用名、FDA分類名、產(chǎn)品貿(mào)易名;
5) 企業(yè)注冊號碼,如企業(yè)在遞交510(K)時已進行企業(yè)注冊,則應(yīng)給出注冊信息,若未注冊,也予注明;
6) 分類,即產(chǎn)品的分類組、類別、管理號和產(chǎn)品代碼;
7) 性能標準,產(chǎn)品所滿足的強制性標準或自愿性標準;
8) 產(chǎn)品標識,包括包裝標識、使用說明書、包裝附件、產(chǎn)品標示等;
9) 實質(zhì)相等性比較(SE);
10) 510(K)摘要或聲明;
11) 產(chǎn)品描述,包括產(chǎn)品的預(yù)期用途、工作原理、動力來源、零組件、照片、工藝圖、裝配圖、結(jié)構(gòu)示意圖等;
12) 產(chǎn)品的安全性與有效性,包括各種設(shè)計、測試資料;
13) 生物相容性;
14) 色素添加劑(如適用);
15) 軟件驗證(如適用);
16) 滅菌(如適用),包括滅菌方法的描述、滅菌驗證產(chǎn)品包裝和標識等。
510(K)申請通過審核后,FDA發(fā)出批準函件。
FDA官方費用(2024年)
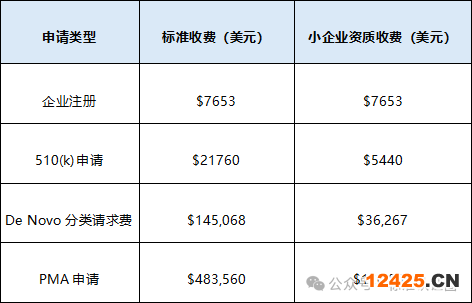
申請資料
510(k)提交:eSTAR及其附件。
PMA提交:eCopy,包括更為詳盡的臨床數(shù)據(jù)和研究報告。
注冊流程
確定產(chǎn)品是否屬于醫(yī)療器械及風(fēng)險分類510(k)(上市前通知)
指定美國代理人
準備注冊文件
提交注冊文件至FDA
獲取醫(yī)療器械注冊證書
合法進入美國市場銷售
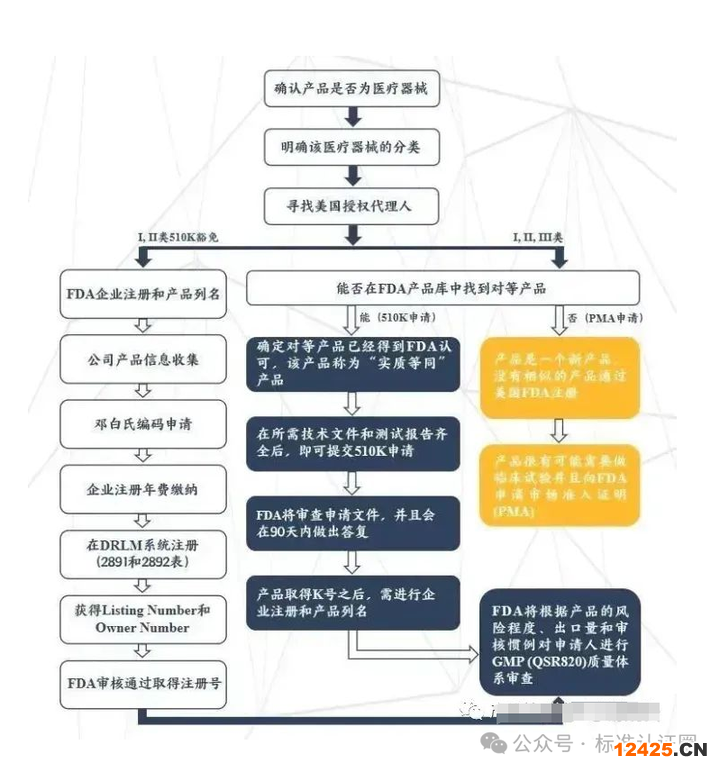
審批時間
注冊時間根據(jù)產(chǎn)品風(fēng)險等級分類的不同,從幾周到幾個月不等。
中企檢測認證網(wǎng)提供iso體系認證機構(gòu)查詢,檢驗檢測、認證認可、資質(zhì)資格、計量校準、知識產(chǎn)權(quán)貫標一站式行業(yè)企業(yè)服務(wù)平臺。中企檢測認證網(wǎng)為檢測行業(yè)相關(guān)檢驗、檢測、認證、計量、校準機構(gòu),儀器設(shè)備、耗材、配件、試劑、標準品供應(yīng)商,法規(guī)咨詢、標準服務(wù)、實驗室軟件提供商提供包括品牌宣傳、產(chǎn)品展示、技術(shù)交流、新品推薦等全方位推廣服務(wù)。這個問題就給大家解答到這里了,如還需要了解更多專業(yè)性問題可以撥打中企檢測認證網(wǎng)在線客服13550333441。為您提供全面檢測、認證、商標、專利、知識產(chǎn)權(quán)、版權(quán)法律法規(guī)知識資訊,包括商標注冊、食品檢測、第三方檢測機構(gòu)、網(wǎng)絡(luò)信息技術(shù)檢測、環(huán)境檢測、管理體系認證、服務(wù)體系認證、產(chǎn)品認證、版權(quán)登記、專利申請、知識產(chǎn)權(quán)、檢測法、認證標準等信息,中企檢測認證網(wǎng)為檢測認證商標專利從業(yè)者提供多種檢測、認證、知識產(chǎn)權(quán)、版權(quán)、商標、專利的轉(zhuǎn)讓代理查詢法律法規(guī),咨詢輔導(dǎo)等知識。
本文內(nèi)容整合網(wǎng)站:百度百科、搜狗百科、360百科、知乎、市場監(jiān)督總局 、國家認證認可監(jiān)督管理委員會、質(zhì)量認證中心
免責(zé)聲明:本文部分內(nèi)容根據(jù)網(wǎng)絡(luò)信息整理,文章版權(quán)歸原作者所有。向原作者致敬!發(fā)布旨在積善利他,如涉及作品內(nèi)容、版權(quán)和其它問題,請跟我們聯(lián)系刪除并致歉!



